The problem “subs - Finding a Motif in DNA” has the assignment to find all matches of a motif in a given string. The sample dataset is as followed:
Sample input
GATATATGCATATACTT
ATAT
Sample output
2 4 10
In theory, I have learned different algorithms on how to solve such problems. Now I want to apply them to this (and eventually bigger) datasets.
I will discuss four different algorithms to find motifs in a DNA sequence:
- Naive Approach - Pattern Matching Problem
- KMP: Knuth-Morris-Pratt
- Rabin-Carp
- Bozer-Moore
Load data
Even though these strings are really fast copied and pasted into my IDE, I like to always consider how this would be possible (especially with Rosalind’s time limit of 5 minutes) when the data is much larger in size or quantity. Then command+C and command+V is not the best option; it just takes too much time and RAM.
f = open('rosalind_subs.txt', 'r')
subsList = f.read().split()
s = subsList[0]
t = subsList[1]
So I used the functions open(), read() and then split() to get the strings from the downloaded dataset. In this case you do not need to split the txt file in any complicated way, but just by line separation. This splits the text in multiple (here just 2) strings and stores them in a list. The DNA and the Motif can be received by their indices.
Here, what is finally stored in both variables s and t:
print('DNA (s):', s)
print('\nMotif to find (t):',t)
DNA (s): CCAGTGGGGTTAGTGCAGTGGGCAGTGGGCAGTGGGGGACGCTGAGCGTCAGTGGGCAGTGGGGGCCATAATGAAACGTGTCACATGCAAATACAGTGGGGGAGCAGTGGGCAGTGGGCCAGTGGGCAGTGGGTGGGACCAGTGGGCAGTGGGGTATTCAGTGGGGCAGTGGGCAGTGGGCAGTGGGAATTCAAATTCAGTGGGCAGTGGGCAGTGGGCTGCCTTCCCAGTGGGCAGTGGGATGCCAGTGGGCCCAGTGGGTAGGCAGTGGGTCAGTGGGGACTCAGTGGGCGCAGTGGGACCAGTGGGCAGTGGGCACAGTGGGAAGGCAGTGGGAAGCAGTGGGCACAGTGGGCTTTCCAGTGGGTCAGTGGGCGGTCACAGTGGGCAGTGGGCAGTGGGTCAGTGGGGGGCAGTGGGGCAGTGGGTACAGTGGGGAACCAGTGGGTTCCTATCCACGTCAGTGGGCAGTGGGACAGTGGGGGCCAGTGGGCAGTGGGCAGTGGGCAGTGGGTATTTCCAGTGGGCAGTGGGCAGTGGGTCAGTGGGATCAGGCAGTGGGTATCCCCTATGCAGCAGCGGCAGCAGTGGGCAATTCTCAGTGGGTAGCGGGCGCGGTGCAGTGGGCAGTGGGTGCAGTGGGGTGCAGTGGGATCAGTGGGCTCACGTCAGTGGGCCCAGTGGGCATAGGTGTGTACAGTGGGACAGTGGGCAGTGGGCAGACAGTGGGACAGTGGGCAGTGGGGCAGTGGGCAGTGGGTCGCAGTGGGTGCAGTGGGCTGGCAGTGGGCAGTGGGCCAGTGGGACTTCAGTGGG
Motif to find (t): CAGTGGGCA
You can prepare this before actually getting the first (large) dataset and before you can simply practice with the small sample dataset.
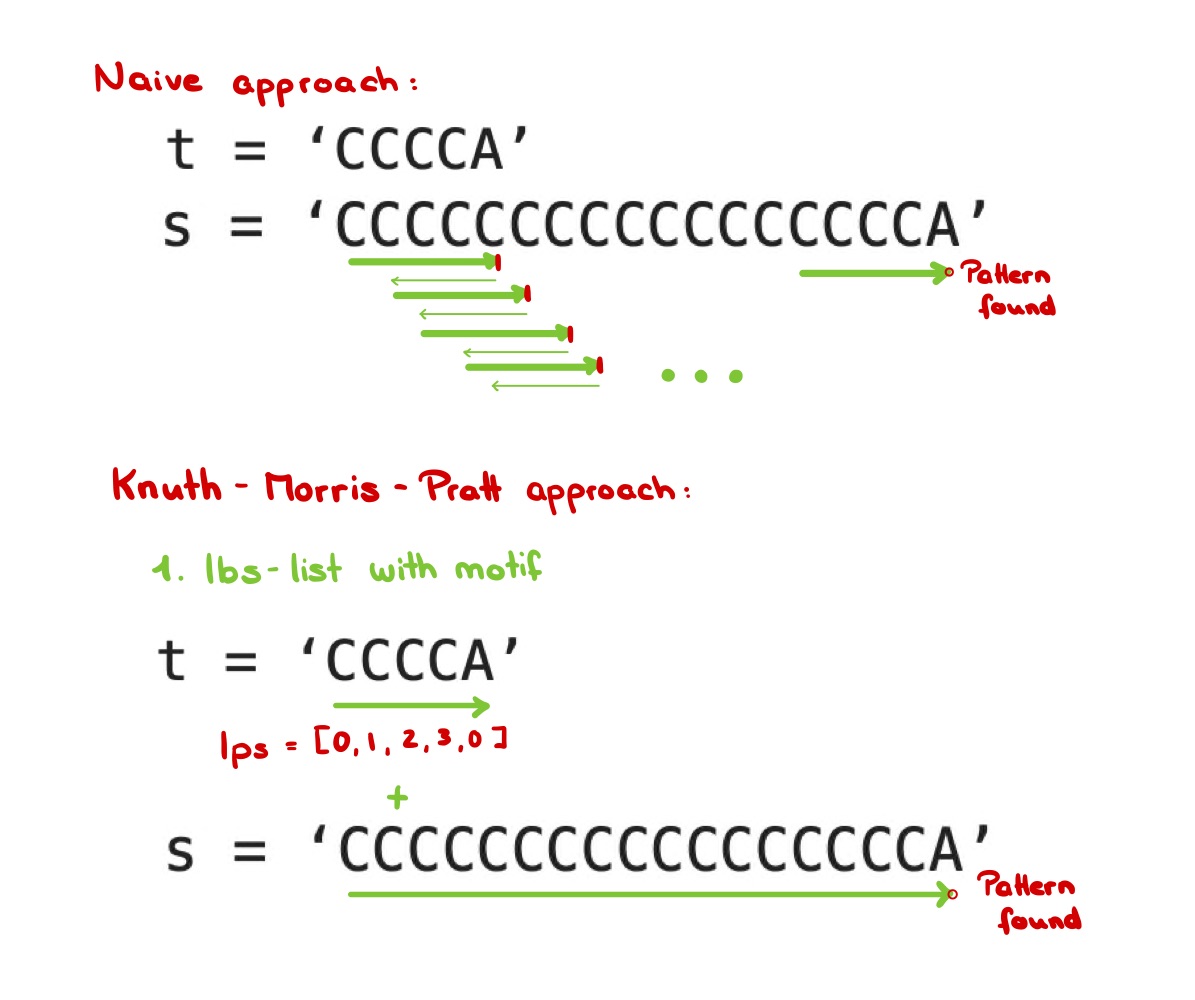
1. Naive Approach
Time complexity: O (nm)
Here, n represents the length of the complete DNA string and m for the length of the Motif.
Following my function that can be called with naive_search(s,t):
def naive_search(DNA, motif):
loc = set() #set for all starting-locations of motifs>
dnaShort = len(DNA) - len(motif) + 1>
for pos in range(dnaShort):
counter = 0 #counting matches between DNA sequence and Motif
for nucl in range(len(motif)):
if motif[nucl] == DNA[pos + nucl]:
counter += 1
else:
break
if counter == len(motif):
pos += 1
loc.add(pos)
#print(motif, "found at position", pos)
loc = sorted(loc)
print('Output:', *loc)
Even though the main focus of this post are different ways solving this problem, still following an explanation of a couple of lines:
Line 3 is built to change the value of in the range value in to prevent an “IndexError” in line 6.

However, this can cause errors in line 7 when coming to the end of the DNA sequence s:

How to solve this? One approach is just to insert an exception (try-except). Or instead, end the for-loop in line 4 earlier. I created dnaShort-value in line 3 for this purpose:

On another note: Generally Rosalind does not seem to like the answers in lists (or sets), so in line 17, you can find a great way to easily separate each value of the list with a space. Just add a star before the variable. Quite straight forward.
Check out the latest code on GitHub: Commit to Naive Approach